Vulnerability of neurons
The “regional” character of neurodegeneration in neurodegenerative diseases, such as Huntington’s disease (striatum and cortex) and Parkinson’s disease (substantia nigra pars compacta) remains unexplained. Our working hypothesis is that the
vulnerability of the various neurons affected in neurodegenerative diseases results from complex mechanisms involving both the cellular “context” and the intrinsic characteristics of the cells or their “phenotype”. We have addressed this hypothesis through many studies in the recent years. We aim at discovering new factors/molecular mechanisms regulating the dysfunction and death of neurons in a loco-regional manner in neurodegenerative diseases. This could help to identify new therapeutic targets. In parallel, it also contributes to develop novel
genetic models and assess cutting-edge brain imaging methods (MRI, NMR spectroscopy, PET) to characterize these models.
Huntington's disease
In particular, our findings support the idea that damage to the mitochondrial/energy system is responsible for the vulnerability of the striatum for Huntington's disease (HD) (Brouillet et al., 1995; Benchoua et al, 2006&2008, Damiano et al., 2013). Interestingly we showed that the presence of dopamine in the striatum, an integral part of the cell "context", renders striatal neurons more vulnerable to the toxicity of huntingtin via their D2-type membrane receptors, which are key molecular markers of striatal neurons (phenotype). On one hand, these results led us to study the role of newly identified molecular markers of striatal neurons in Huntington's disease pathogenesis (for a review: Francelle et al., 2014).
We determined how a short list of molecular markers of the striatum, we identified by a genetic screen in 2008, and whose function was largely unknown could act as "disease modifiers" in Huntington's disease. For this purpose, we used different genetic models of Huntington's disease and viral vector gene transfer methods, to assess how knocking down or overexpressing these markers (Gpr88, Crym, Capucin, DCLK3, and the long non-coding RNA ABHD11-AS) (Galvan et al, 2012, Francelle et al., 2014, 2015).
This work is now focusing on the kinase DCLK3 which expression is reduced early in Huntington's disease. We have started to decipher its potential roles in the striatum and found it plays a protective role in medium size spiny neurons of the striatum through mechanisms that likely involve its interaction with a nuclear complex regulating transcription. A manuscript on DCLK3 is under revision. For the DCLK3 project, the FRM awarded a Doctorate fellowship to Lucie de Longprez in 2015. Recently, our group obtained an ANR grant in 2017 as partner (EpIHD, coordinated by Karine Merienne, Univ. Strasbourg) focusing on epigenetic deregulations in Huntington's disease and the possible implication of DCLK3 in epigenetic regulations.
On the other hand, we continue to explore how metabolic defects underlie Huntington's disease metabolism. For this we study mouse models (KI140CAG) and BACHD rats using novel MRI and NMR spectroscopy approaches. This program is supported by a grant from ANR and DGOS (ANR "HDeNERGY" 2014-2018, coordinator Emmanuel Brouillet). NMR methods develop in animal models are translated to the clinic thanks to our collaboration with the ICM/Department of Genetics, (Dr Fanny Mochel, Salpêtrière Hospital, Paris). We identify new biomarkers that could be used to assess the efficacy of therapeutic treatments.
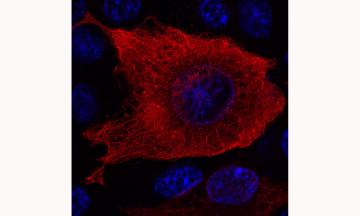
Cellular distribution of the striatal protein DCLK3. The recombinant DCLK3 protein was generated by transfecting human cells and was detected by immunofluorescence and visualized by confocal microscopy. The DNA of the nuclei is shown in blue. DCLK3 can be seen to form a network in the cytoplasm, consistent with its ability to interact with microtubules.
Parkinson's disease
We develop
new models of Parkinson's disease based on viral vectorization (lentivirus, adenoassociated virus). to overexpress C-terminal fragments of the
LRRK2 gene in rodents. We are especially interested in two proteins that are likely central in Parkinson's disease pathogenesis, LRRK2 and a-synuclein. Mutations of LRRK2 gene (especially the G2019S substitution) are associated with the most frequent genetic forms of the disease. Two key questions are addressed regarding LRRK2. What is the actual role of its kinase catalytic activity, which is increased compared to the wild type form when the G2019S mutation is present? How could the presence of LRRK2 modulate the toxicity of a-synuclein ? We found that when expressed at high level the C-ter-LRRK2 constructs harbouring the G2019S mutation produces significant loss of dopaminergic neurons in the substantia nigra pars compacta. This effect is not seen with the wild type form of LRRK2. This demonstrates that the toxicity of LRRK2 toward dopaminergic neurons could at least in part result from an increase in its kinase activity, independently of the N-terminal domain. We also showed that the toxicity of a-synuclein is significantly increased by the presence of C-ter-LRRK2-G2019S (at early time points when C-ter-LRRK2-G2019S alone is not toxic per se).
In contrast, the wildtype form of the protein is not toxic. Interestingly, the dead kinase mutant of C-ter-LRRK2-G2019S (D1994A) does not change a-synuclein toxicity, further supporting the hypothesis that there exists a functional interaction between LRRK2 and a-synuclein and that the increased kinase activity of Cter-LRRK2 is a key mechanisms modulating a-synuclein neurotoxicity in dopaminergic neurons
in vivo. These data have been mainly obtained during Noémie Cresto's thesis. Noémie Cresto is recipient of a grant form "Association France Parkinson".
The entire program to develop the viral vector-based models has been supported by grants awarded by "Fondation de France" (Parkinson Committee 2011-2016).
We now pursue this project with two directions:
- One is to decipher the underlying mechanisms of the synergy between LRRK2 and a-synuclein.
Dr
Géraldine Liot is in charge of this project. For details see (link).
- We also pursue the development of Parkinson's disease models using AAVs expressing different forms of a-synuclein and LRRK2 in the context of a European Initial Training Network Marie S. Curie project (Coordinator Abhay Pandit, Gallway, Ireland) called "BrainMatTrain" (see
http://curamdevices.ie/curam/research/eu-projects/brainmattrain/).
This exciting project aims at assessing the possibility of using novel biomaterial (hydrogels) that could be functionalized to slowly release neuroprotective agents after injection into the brain in models of Parkinson's disease. Francesco Gubinelli is working as a PhD student of this international program in the lab since December 2016.
Members of the laboratory associated with these projects
- Noemie Cresto (PhD; CEA and Association France Parkinson): Defended her PhD in July 2017
- Lucie de Longprez (PhD student; CEA and FRM): "DCLK3, a new striatal marker conferring neuroprotection in the context of Huntington's disease"
- Francesco Gubinelli (PhD student; ITN Marie S. Curie) : "Long-term functional neuronal repair by multi-modal biomaterial system in vivo models of PD"
- Maria-Angeles Carillo de Sauvage:
maria.carrillo@cea.fr
- Marie-Claude Gaillard (CEA Engineer):
marie-claude.gaillard@cea.fr
- Caroline Jan (CNRS Engineer):
caroline.jan@cea.fr
- Martine Guillermier (CNRS CNRS Engineer):
martine.guillermier@cea.fr
- Julien Flament (INSERM Ingeneer, US27):
Julien.flament@cea.fr
Collaborations
- Pr M. Flint Beal, Department of Neurology and Neuroscience, Weill Cornell Med College, Cornell University, NY, USA.
- Prof. Dr. Tiago Fleming Outeiro, Department of NeuroDegeneration and Restorative Research
- Center of Molecular Physiology of the Brain, University Medizin Goettingen, Germany.
- Pr Nicole Déglon, Laboratoire des neurothérapies cellulaires et moléculaires, CHUV, Lausanne, Suisse.
Recent Reviews
Imaging and spectroscopic approaches to probe brain energy metabolism dysregulation in neurodegenerative diseases
Bonvento G, Valette J, Flament J, Mochel F, Brouillet E.
J Cereb Blood Flow Metab. 2017, 37(6):1927-1943. Review.
Contribution of Neuroepigenetics to Huntington's Disease
Francelle L, Lotz C, Outeiro T, Brouillet E, Merienne K.
Front Hum Neurosci. 2017, 11:17. Review.
Energy defects in Huntington's disease: Why "in vivo" evidence matters
Liot G, Valette J, Pépin J, Flament J, Brouillet E.
Biochem Biophys Res Commun. 2016 Sep 14.
Possible involvement of self-defense mechanisms in the preferential vulnerability of the striatum in Huntington's disease
Francelle L., Galvan L., Brouillet E.
Frontiers in Cellular Neurosciences, 2014, 8:295.
Recent original publications
The Self-Inactivating KamiCas9 System for the Editing of CNS Disease Genes
Merienne N, Vachey G, de Longprez L, Meunier C, Zimmer V, Perriard G, Canales M, Mathias A, Herrgott L, Beltraminelli T, Maulet A, Dequesne T, Pythoud C, Rey M, Pellerin L, Brouillet E, Perrier AL, du Pasquier R, Déglon N.
Cell Rep. 2017, 20(12):2980-2991.
Altered enhancer transcription underlies Huntington's disease striatal transcriptional signature
Le Gras S, Keime C, Anthony A, Lotz C, De Longprez L, Brouillet E, Cassel JC, Boutillier AL, Merienne K. .
Sci Rep. 2017, 7:42875.
In vivo imaging of brain glutamate defects in a knock-in mouse model of Huntington's disease
Pépin J, Francelle L, Carrillo-de Sauvage MA, de Longprez L, Gipchtein P, Cambon K, Valette J, Brouillet E, Flament
J. Neuroimage. 2016 Jun 16;139:53-64.
New paradigm to assess brain cell morphology by diffusion-weighted MR spectroscopy
in vivo
Palombo M, Ligneul C, Najac C, Le Douce J, Flament J, Escartin C, Hantraye P, Brouillet E, Bonvento G, Valette J. (2016)
Proc Natl Acad Sci U S A. 2016, 113(24):6671-6.
Synaptic scaling up in medium spiny neurons of aged BACHD mice: A slow-progression model of Huntington's disease
Rocher AB, Gubellini P, Merienne N, Boussicault L, Petit F, Gipchtein P, Jan C, Hantraye P, Brouillet E, Bonvento G.
Neurobiol Dis. 2016, 86:131-9.
The striatal long non-coding RNA Abhd11os is neuroprotective against an N-terminal Fragment of Mutant Huntingtin
in vivo
Francelle L*, Galvan L *, Gaillard MC, Petit F, Bernay B, Guillermier M, Bonvento G, Dufour N, Elalouf JM, Hantraye P, Déglon N, de Chaldée M, and Brouillet E.
Neurobiol Aging, 2015, 36(3) (*, co-first authors)
Loss of the thyroid hormone binding protein Crym renders striatal neurons more vulnerable to mutant huntingtin in Huntington's disease
Francelle L, Galvan L, Gaillard MC, Guillermier M, Houitte D, Bonvento G, Petit F, Jan C, Dufour N, Hantraye P, Elalouf JM, de Chaldée M, Déglon N, Brouillet E.
Hum Mol Genet. 2015, 2015 24(6):1563-73. (*, co-first authors)
Impaired brain energy metabolism in the BACHD mouse model of Huntington's disease: critical role of astrocyte-neuron interactions
Boussicault L, Hérard AS, Calingasan N, Petit F, Malgorn C, Merienne N, Jan C, Gaillard MC, Lerchundi R, Barros LF, Escartin C, Delzescaux T, Mariani J, Hantraye P, Flint Beal M, Brouillet E, Véga C, Bonvento G.
J Cereb Blood Flow Metab. 2014 Jun 18.
A role of mitochondrial complex II defects in genetic models of Huntington's disease expressing N-terminal fragments of mutant huntingtin
Damiano M, Diguet E, Malgorn C, D'Aurelio M, Galvan L, Petit F, Benhaim L, Guillermier M, Houitte D, Dufour N, Hantraye P, Canals JM, Alberch J, Delzescaux T, Déglon N, Beal MF, Brouillet E
Hum Mol Genet.2013 22:3869-82.
Capucin does not modify the toxicity of a mutant Huntingtin fragment in vivo
Galvan L, Lepejová N, Gaillard MC, Malgorn C, Guillermier M, Houitte D, Bonvento G, Petit F, Dufour N, Héry P, Gérard M, Elalouf JM, Déglon N, Brouillet* E, de Chaldée* M.
Neurobiol Aging., 2012 - 1845.e5-6.